Syndrome de Klein-Waardenburg : Comprendre ce sous-type des syndromes de Waardenburg et ses caractéristiques cliniques

Le syndrome de Klein-Waardenburg est une maladie génétique rare qui fait partie du groupe des syndromes de Waardenburg. Ce trouble se caractérise par une combinaison d’anomalies de la pigmentation de la peau, des cheveux et des yeux, ainsi que par une perte auditive congénitale. Ce syndrome intègre des caractéristiques propres au syndrome de Waardenburg, avec des anomalies physiques additionnelles qui lui confèrent une identité particulière. Dans cet article, nous allons explorer en profondeur le syndrome de Klein-Waardenburg, ses caractéristiques cliniques, sa génétique, les diagnostics et les possibilités de traitement, tout en répondant à certaines questions fréquemment posées sur cette pathologie.
Qu’est-ce que le syndrome de Klein-Waardenburg?
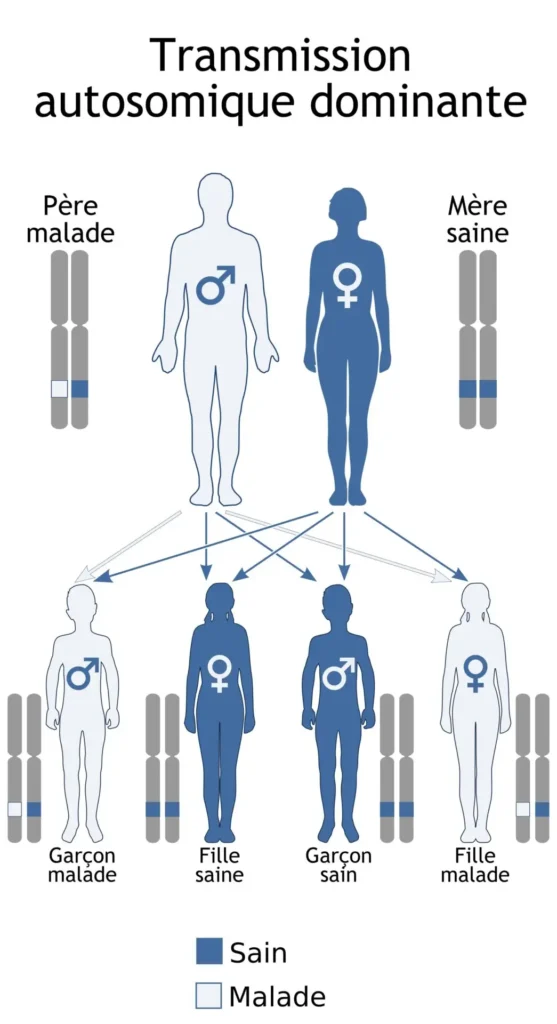
Le syndrome de Klein-Waardenburg est un sous-type du syndrome de Waardenburg, décrit pour la première fois par Waardenburg en 1951. Il s’agit d’un trouble autosomique dominant, c’est-à-dire qu’une seule copie du gène mutant, hérité d’un parent, suffit à provoquer la maladie. Parmi les caractéristiques principales de ce syndrome, on retrouve une perte auditive congénitale présente chez environ 60 % des patients, ainsi que des anomalies de la pigmentation, qui se manifestent généralement par des mèches de cheveux blanches (poliose), une hétérochromie (couleurs différentes entre les deux yeux) ou une peau dépigmentée.
Le syndrome de Klein-Waardenburg se distingue des autres formes par des anomalies musculo-squelettiques marquées, notamment au niveau des membres supérieurs. Les individus atteints présentent souvent une hypoplasie des bras, des contractures en flexion, une fusion des os du carpe, ainsi qu’une syndactylie. On observe également une aplasie des deux premières côtes et parfois une dystopie des clavicules. Ce syndrome inclut des caractéristiques faciales distinctives, telles qu’une synophris (sourcils jointifs), un élargissement de la base du nez, des canthus dystopiques, et des blépharophimosis. La peau et les cheveux montrent des signes d’albinisme partiel, les yeux peuvent présenter une hétérochromie irienne bleue, et la surdité, parfois accompagnée de mutité, est fréquente.
Types de syndrome de Klein-Waardenburg
Le syndrome de Klein-Waardenburg peut être classé en quatre sous-types en fonction de la présence et de la gravité des symptômes. Ces types sont définis par des variations dans les mutations génétiques et les manifestations cliniques. Les principaux types incluent :
Syndrome de Waardenburg de Type 1 (WS 1)
Les principales caractéristiques du type 1 du syndrome de Waardenburg incluent :
- Ectopie : déplacement latéral des angles palpébraux médians, entraînant un large espace entre les yeux.
- Hétérochromie irienne : les iris des deux yeux sont souvent de couleurs différentes.
- Présence de mèches de cheveux blanches et de duvet clair.
- Surdité neurosensorielle : perte auditive pouvant varier en sévérité.
Syndrome de Waardenburg Type 2 (WS 2)
Le type 2 du syndrome de Waardenburg se distingue par une fréquence plus élevée de surdité congénitale, touchant environ 50 % des individus. Comme dans le type 1, il présente des anomalies de pigmentation des cheveux, de la peau et des yeux, mais sans ectopie. Ce type se caractérise également par des anomalies plus marquées de la pigmentation ainsi qu’une hypoplasie notable des membres supérieurs, accompagnée de particularités craniofaciales qui renforcent les signes distinctifs du syndrome.
Syndrome de Waardenburg Type 3 (WS 3 ou Klein-Waardenburg)
Le type 3, également appelé syndrome de Klein-Waardenburg, présente des similarités avec les deux premiers types, notamment la perte d’audition et des anomalies pigmentaires, ainsi que des traits faciaux distinctifs comme un nez large et des yeux espacés. Ce type se distingue par des malformations musculo-squelettiques marquées, touchant les membres supérieurs, avec des faiblesses au niveau des bras ou des épaules et des malformations articulaires. Dans certains cas, une dystopie des clavicules peut également être observée, accentuant la sévérité des atteintes musculo-squelettiques.
Syndrome de Waardenburg Type 4 (WS 4 ou Shah-Waardenburg)
Le type 4, connu sous le nom de syndrome de Waardenburg-Shah, est une forme particulière du syndrome de Waardenburg caractérisée par des anomalies pigmentaires et une possible perte d’audition. Contrairement aux autres types, il se transmet de manière autosomique récessive et est associé à la maladie de Hirschsprung, une affection pouvant provoquer des constipations sévères et des blocages intestinaux en raison d’une absence de cellules nerveuses dans certaines parties de l’intestin.
Prévalence et étiologie
Le syndrome de Klein-Waardenburg est extrêmement rare. On estime qu’il touche environ 1 à 9 personnes sur 1 000 000. Cette pathologie résulte de mutations génétiques qui affectent principalement le gène PAX3 situé sur le chromosome 2q35. Ce gène joue un rôle crucial dans le développement embryonnaire, en particulier pour la formation des cellules mélanocytaires, responsables de la pigmentation, ainsi que des cellules nerveuses de la crête neurale. Une altération de ce gène peut provoquer des anomalies dans la formation de la structure de la peau, des yeux et de l’ouïe.
Symptômes et caractéristiques cliniques du syndrome de Klein-Waardenburg
Les personnes atteintes du syndrome de Waardenburg type 3 présentent une gamme de symptômes qui peuvent varier en sévérité. Les manifestations typiques incluent :
- Surdité congénitale : Celle-ci peut être uni ou bilatérale, et de degré variable, allant de la surdité légère à une surdité totale. La surdité est souvent le premier signe qui alerte les médecins.
- Anomalies pigmentaires : L’une des caractéristiques les plus visibles est l’hétérochromie (deux yeux de couleurs différentes). Une mèche de cheveux blanche ou des taches cutanées dépigmentées sont également des signes distinctifs.
- Anomalies des membres : Des déformations des bras et des mains sont caractéristiques du type 3. Cela peut inclure des coudes contractés, des mains en flexion permanente, ou des doigts soudés.
- Retard de développement : Bien que rare, certains cas de type 3 peuvent présenter un retard de développement moteur dû aux anomalies musculo-squelettiques.
Diagnostic du syndrome de Klein-Waardenburg
Le diagnostic du syndrome de Klein-Waardenburg se fait principalement sur la base des signes cliniques observables chez le patient et de l’histoire familiale. Les médecins utilisent souvent un algorithme basé sur les principaux signes cliniques énumérés ci-dessus, et dans la majorité des cas, le diagnostic peut être confirmé par un test génétique qui identifie des mutations dans le gène PAX3.
En plus de l’évaluation clinique, l’imagerie médicale et des tests auditifs sont souvent nécessaires pour confirmer la présence des anomalies musculo-squelettiques et de la perte auditive. Un conseil génétique est également recommandé pour les familles affectées, afin de les aider à comprendre la nature de la maladie et ses possibilités de transmission.
Pathophysiologie et génétique
Le syndrome de Waardenburg type 3 est un trouble autosomique dominant, ce qui signifie qu’une seule copie du gène muté, transmise par l’un des parents, est suffisante pour causer le syndrome. Cependant, certains cas peuvent aussi être dus à de nouvelles mutations, c’est-à-dire des mutations spontanées qui ne sont pas héritées des parents.
Les mutations responsables du syndrome de Waardenburg touchent principalement les gènes PAX3 et MITF, qui jouent un rôle crucial dans le développement des cellules de la crête neurale. Pour le type 3, des mutations spécifiques du gène PAX3 sont souvent impliquées, ce qui explique les malformations musculo-squelettiques qui sont caractéristiques de ce type.
Options de traitement
Il n’existe actuellement aucun traitement curatif pour le syndrome de Klein-Waardenburg. Le traitement est principalement symptomatique et vise à améliorer la qualité de vie des personnes atteintes.
- Correction de la perte auditive : Les appareils auditifs et les implants cochléaires peuvent être utilisés pour améliorer l’audition des patients. Le choix de l’appareil dépend du type et de la gravité de la perte auditive.
- Thérapies de rééducation : Les patients peuvent bénéficier de différents types de thérapies, comme la rééducation auditive et l’orthophonie, qui visent à améliorer la communication.
- Interventions chirurgicales : En cas d’anomalies musculo-squelettiques sévères, des interventions chirurgicales peuvent être nécessaires pour corriger des déformations des membres.
Pronostic et qualité de vie
Le pronostic des personnes atteintes du syndrome de Klein-Waardenburg est généralement bon, surtout lorsqu’une prise en charge précoce est mise en place pour réduire les conséquences de la perte auditive et des anomalies physiques. Cependant, la qualité de vie des patients peut être affectée par les défis liés à la communication et aux stigmates sociaux associés aux anomalies physiques.
Conseil génétique et prévention
Le conseil génétique est très important pour les familles présentant des antécédents de syndrome de Klein-Waardenburg. Les professionnels de santé peuvent aider à évaluer le risque de transmission de la maladie aux futures générations et discuter des options disponibles, comme le diagnostic préimplantatoire pour les couples souhaitant avoir des enfants non affectés.
Recherche actuelle et perspectives d’avenir
La recherche sur le syndrome de Klein-Waardenburg est en cours, notamment dans les domaines de la génétique et des thérapies géniques potentielles. Des approches innovantes, telles que la manipulation de gènes ou l’utilisation de cellules souches pour réparer les tissus affectés, sont en cours d’investigation. Bien que ces approches soient encore au stade expérimental, elles offrent un espoir pour l’avenir des patients atteints de troubles génétiques rares.
Conclusion
Le syndrome de Klein-Waardenburg est une pathologie rare qui affecte différents aspects de la vie des personnes atteintes, en raison de la perte auditive et des anomalies de la pigmentation et du squelette. Bien qu’il n’existe pas de traitement curatif, une prise en charge précoce et multidisciplinaire permet d’améliorer considérablement la qualité de vie des patients. La recherche continue sur cette maladie pourrait offrir de nouvelles perspectives thérapeutiques dans le futur.
FAQ sur le Syndrome de Waardenburg
1. Qu’est-ce que le syndrome de Waardenburg ?
Le syndrome de Waardenburg est un groupe de syndromes rares caractérisés par des troubles de la pigmentation, des anomalies cranio-faciales et une perte auditive congénitale.
2. Quelles sont les caractéristiques du syndrome de Klein-Waardenburg ?
Le syndrome de Klein-Waardenburg, un sous-type du syndrome de Waardenburg, présente des dysmorphies faciales, des anomalies des membres, ainsi qu’une perte auditive et des troubles de la pigmentation, comme une mèche blanche frontale.
3. Comment se manifeste la dysmorphie faciale dans ces syndromes ?
Les patients peuvent présenter des traits distinctifs tels qu’une racine du nez large et une dystopie des canthi internes des yeux, contribuant à l’aspect facial unique.
4. Quelles anomalies sont associées aux membres ?
Des anomalies des membres peuvent inclure des malformations, telles que des doigts supplémentaires (polydactylie) ou des déformations articulaires.
5. Quels types de troubles de la pigmentation sont observés ?
Les troubles de la pigmentation incluent l’hétérochromie (couleurs différentes des yeux) et la poliose (mèche blanche dans les cheveux).
6. Quelle est la transmission génétique de ces syndromes ?
Ces syndromes se transmettent par un mode autosomique dominant, ce qui signifie qu’une seule copie du gène muté suffit pour provoquer la maladie.
7. Quelles sont les différences entre les types 1 et 2 du syndrome de Waardenburg ?
Les types 1 et 2 se distinguent principalement par la sévérité des anomalies de pigmentation et les malformations musculo-squelettiques, avec le type 1 présentant généralement moins d’anomalies.
8. Quels sont les risques associés au syndrome rare du Klein-Waardenburg ?
Les individus atteints peuvent souffrir d’une perte auditive significative et d’anomalies physiques qui peuvent affecter leur qualité de vie.
9. Comment se déroule le diagnostic ?
Le diagnostic repose sur l’évaluation clinique des symptômes, l’historique familial et peut être confirmé par des tests génétiques pour identifier les mutations associées.
10. Quelles sont les options de traitement disponibles ?
Bien qu’il n’existe pas de traitement curatif, les options incluent des appareils auditifs, des interventions chirurgicales pour corriger les malformations et un suivi régulier pour gérer les symptômes.
Sources utilisées
- Revue de médecine périnatale, 2017 – « Syndrome de Klein-Waardenburg » Disponible ici
- GeneReviews – « Waardenburg Syndrome Types I-IV Overview »
- Orphanet – « Waardenburg syndrome type III »
- National Center for Biotechnology Information (NCBI) – Publications sur les mutations du gène PAX3
- « Syndrome de Waardenburg », Amplifon