
Le Syndrome d’Alström : Symptômes, Causes et traitements d’une maladie rare

Le syndrome d’Alström est une maladie génétique très rare, également décrite comme une pathologie multisystémique héréditaire qui affecte plusieurs organes du corps. Ce syndrome a été découvert pour la première fois en 1959 par le docteur Carl-Henry Alström. Étant une condition héréditaire autosomique récessive, il est nécessaire que les deux parents soient porteurs du gène muté pour que leur enfant présente la maladie. Bien que rare, le syndrome présente de nombreux signes cliniques et des complications importantes qui peuvent apparaître dès l’enfance.
Dans cet article, nous aborderons en détail les symptômes, les causes, le diagnostic, les complications, et les options de traitement disponibles pour le Syndrome d’Alström. Nous allons examiner en profondeur cette maladie complexe pour mieux comprendre comment elle impacte la vie des patients et de leurs familles.
Qu’est-ce que le syndrome d’Alström ?
Le syndrome d’Alström est une maladie génétique très rare qui touche divers systèmes corporels. Les patients présentent typiquement une dystrophie des cônes et des bâtonnets dans la rétine, une surdité progressive, de l’obésité, une résistance à l’insuline, ainsi que des problèmes cardiaques et pulmonaires. Il s’agit d’une maladie multisystémique, ce qui signifie qu’elle affecte plusieurs organes et fonctions du corps humain.
Signes et symptômes du syndrome d’Alström
Les symptômes du Syndrome d’Alström peuvent varier significativement d’une personne à l’autre, et ils apparaissent généralement dès la petite enfance. Parmi les principaux symptômes, on trouve :
- Problèmes de vision : La dystrophie des cônes et des bâtonnets affecte précocement la vision en entraînant une perte progressive de la vision périphérique, puis centrale. Ce trouble, pouvant évoluer en rétinite pigmentaire à cônes, provoque des symptômes comme un nystagmus (mouvements saccadés et involontaires des yeux) et une photophobie marquée, rendant les patients sensibles à la lumière. Dans les cas graves, cette dégénérescence peut entraîner une cécité dès l’adolescence.
- Surdité : La surdité est un autre symptôme fréquent, qui s’aggrave avec le temps et est souvent diagnostiquée dès les premières années de vie. Elle est due à des problèmes sensoriels et neuraux.
- Cardiomyopathie : L’un des signes précoces du Syndrome d’Alström peut être une cardiomyopathie, présentée sous forme de décompensation cardiaque, qui survient parfois dans les premières semaines de la vie d’un nourrisson.
- Obésité et troubles métaboliques : Les patients présentent souvent une obésité qui apparaît très tôt dans l’enfance. Ils sont également prédisposés à développer un diabète de type 2 et une résistance à l’insuline.
- Troubles endocriniens : Les patients peuvent présenter des troubles endocriniens tels qu’une dysfonction thyroïdienne, un hypogonadisme (insuffisance de production des hormones sexuelles) et un hyperandrogénisme (production excessive d’hormones androgènes, entraînant de l’acné et une pilosité anormale) chez les femmes.
- Problèmes rénaux et hépatiques : Une dysfonction rénale et hépatique est courante, et les patients peuvent développer une fibrose hépatique et une néphropathie chronique.
- Fibrose pulmonaire : Les patients peuvent également présenter une fibrose pulmonaire, entraînant des difficultés respiratoires et une diminution de la capacité pulmonaire.
- Scoliose et troubles musculosquelettiques : Des problèmes musculosquelettiques tels que la scoliose (courbure anormale de la colonne vertébrale) peuvent également survenir chez les patients.
- Retard du développement : Certains patients peuvent présenter des retards de développement moteur et cognitif, ainsi que des difficultés d’apprentissage.
- Problèmes gastro-intestinaux : Des troubles gastro-intestinaux, tels que le reflux gastro-œsophagien, des douleurs abdominales chroniques et des troubles de la motilité intestinale, peuvent également être présents.
Les individus atteints du Syndrome d’Alström présentent souvent des caractéristiques physiques distinctes qui peuvent aider au diagnostic. Ces caractéristiques incluent généralement une obésité infantile précoce, un visage rond, un cou court, ainsi qu’une petite taille par rapport à la moyenne. On observe également des doigts et des orteils courts et larges (brachydactylie). Les patients peuvent aussi développer une scoliose, donnant lieu à une courbure anormale de la colonne vertébrale, et leur peau peut présenter des signes de fibrose ou une hyperpigmentation dans certaines zones.
Causes génétiques du syndrome d’Alström
Le Syndrome d’Alström est causé par des mutations dans le gène ALMS1, localisé sur le chromosome 2 (2p13.1). Ce gène joue un rôle essentiel dans la structure et le fonctionnement des cils cellulaires, qui sont importants pour le développement de nombreux tissus et organes du corps. Ces cils sont essentiels pour la signalisation intercellulaire et pour la fonction sensorielle. En raison des mutations du gène ALMS1, les cellules deviennent incapables de répondre correctement aux signaux, ce qui conduit aux divers problèmes cliniques observés chez les patients atteints de ce syndrome.
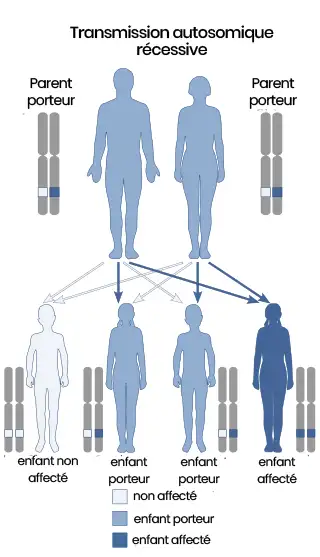
La transmission du syndrome est de type autosomique récessif, ce qui signifie que les deux parents doivent être porteurs du gène mutant pour que l’enfant soit affecté. La probabilité pour qu’un enfant soit atteint si les deux parents sont porteurs est de 25 %.
Diagnostic du syndrome d’Alström
Le diagnostic du syndrome d’Alström est souvent difficile en raison de la grande variété de symptômes et de leur similitude avec d’autres maladies génétiques rares. Le diagnostic précoce est crucial pour une prise en charge optimale, bien que les patients soient souvent diagnostiqués tardivement en raison du manque de spécificité des symptômes.
Le diagnostic repose principalement sur :
- L’observation clinique : La présence de plusieurs des symptômes mentionnés plus haut peut alerter les médecins, en particulier si la combinaison de la cécité, de la surdité et de l’obésité est présente.
- Tests génétiques : La confirmation du diagnostic est obtenue par des tests génétiques pour détecter des mutations dans le gène ALMS1.
- Imageries et examens complémentaires : Des échographies cardiaques et abdominales sont souvent nécessaires pour identifier les complications cardiaques et les atteintes hépatorénales.
Complications associées
Le Syndrome d’Alström est associé à de nombreuses complications graves qui apparaissent au fur et à mesure que la maladie progresse :
- Cardiomyopathie et insuffisance cardiaque : La cardiomyopathie est la principale cause de mortalité chez les personnes atteintes de ce syndrome. L’insuffisance cardiaque peut se manifester tôt dans la vie et s’aggraver avec l’âge.
- Diabète de type 2 : En raison de la résistance à l’insuline, la plupart des patients développent un diabète de type 2 à un âge relativement jeune.
- Problèmes rénaux et hépatiques : Les patients présentent souvent une fibrose hépatique et une dysfonction rénale, augmentant leur vulnérabilité aux infections. Cette dégradation progressive de la fonction rénale peut évoluer vers une insuffisance rénale au stade terminal, nécessitant des soins intensifs pour préserver la qualité de vie.
- Fibrose pulmonaire : Une fibrose pulmonaire entraîne des difficultés respiratoires progressives et diminue la capacité pulmonaire.
Traitement et prise en charge
Il n’existe actuellement aucun traitement curatif pour le syndrome d’Alström. La prise en charge vise principalement à améliorer la qualité de vie des patients et à gérer les symptômes et les complications au fur et à mesure qu’ils apparaissent.
- Traitement médical : Le traitement des complications telles que l’insuffisance cardiaque, le diabète, et les troubles hépato-rénaux est essentiel. Des médicaments tels que les inhibiteurs de l’ECA peuvent être prescrits pour traiter l’insuffisance cardiaque. Pour le diabète, des traitements incluant des antidiabétiques oraux ou de l’insuline peuvent être nécessaires. Des médicaments visant à contrôler l’hypertension et à prévenir les complications rénales sont également souvent utilisés. Des traitements spécifiques pour les maladies pulmonaires, comme les bronchodilatateurs et des anti-inflammatoires, peuvent être prescrits en cas de fibrose pulmonaire.
- Thérapie génétique et traitements expérimentaux : Bien que non encore disponibles pour tous les patients, certaines recherches se concentrent sur le développement de thérapies géniques qui pourraient, à l’avenir, corriger les anomalies génétiques à l’origine du syndrome. Les essais cliniques se multiplient pour explorer l’efficacité de nouveaux traitements expérimentaux visant à améliorer la fonction cellulaire et à ralentir la progression de la maladie.
- Surveillance régulière : Un suivi médical régulier est nécessaire pour évaluer l’évolution de la maladie et ajuster les traitements. Ce suivi inclut des bilans cardiaques, endocriniens, ophtalmologiques et auditifs réguliers. Une surveillance étroite permet d’ajuster les interventions médicales pour chaque patient, en fonction de l’évolution des symptômes.
- Support psychosocial : Les patients et leurs familles ont besoin d’un soutien psychologique et social pour faire face aux effets débilitants de la maladie. Une coordination multidisciplinaire est souvent requise entre différents spécialistes (cardiologue, ophtalmologiste, endocrinologue). Des associations de patients peuvent également fournir des ressources et un soutien précieux pour les familles touchées.
- Aides techniques : Les aides auditives et visuelles, ainsi que les équipements pour la mobilité, peuvent aider à améliorer l’autonomie des patients. Les patients peuvent bénéficier de dispositifs tels que des appareils auditifs, des loupes électroniques, et des fauteuils roulants pour améliorer leur qualité de vie. La mise en place d’un environnement adapté (comme des outils de communication assistée) est souvent essentielle pour garantir l’indépendance des patients.
Perspectives de la recherche
Des efforts sont en cours pour développer de meilleures options de traitement pour le syndrome d’Alström. La recherche se concentre sur la compréhension des mécanismes moléculaires impliqués dans la maladie et sur l’exploration de potentielles thérapies géniques. En outre, des essais cliniques explorent l’utilisation de médicaments qui pourraient atténuer certains des symptômes.
Conclusion
Le syndrome d’Alström est une maladie génétique rare, complexe, et multisystémique qui présente de nombreux défis pour les patients et leurs familles. Bien qu’il n’existe pas encore de remède, la prise en charge multidisciplinaire et un diagnostic précoce peuvent grandement améliorer la qualité de vie des patients. L’éducation et la sensibilisation des médecins et des familles sur les caractéristiques de ce syndrome sont essentielles pour aider à identifier les signes précoces et fournir une prise en charge appropriée.
FAQ sur le Syndrome d’Alström
1. Qu’est-ce que le syndrome d’Alström ?
Le syndrome d’Alström est une maladie génétique très rare, caractérisée par des atteintes multisystémiques, notamment la perte de vision, la surdité et l’obésité.
2. Quels sont les signes de la maladie ?
Les signes incluent une sensibilité à la lumière, une perte auditive neurosensorielle, une cardiomyopathie dilatée, et des dysfonctionnements hépatiques et rénaux.
3. Comment évolue le syndrome d’Alström ?
Le syndrome évolue progressivement, avec des symptômes qui peuvent apparaître dès l’enfance et s’aggraver au fil du temps.
4. Quelle est la prévalence du syndrome d’Alström ?
La prévalence est très faible, estimée à environ 1 cas pour 1 million d’habitants, bien que des taux plus élevés aient été observés dans certaines populations.
5. Quelles complications peuvent survenir ?
Des complications telles que l’insuffisance hépatique, l’insuffisance rénale qui peut conduire à une insuffisance rénale terminale, et des maladies respiratoires chroniques sont courantes.
6. Quel traitement est recommandé pour les patients ?
Le traitement est symptomatique et peut inclure des diurétiques pour gérer l’insuffisance cardiaque congestive ainsi que la prescription de lentilles teintées pour réduire la sensibilité à la lumière.
7. Comment se manifeste la perte auditive dans ce syndrome ?
La perte auditive est généralement progressive et peut varier de légère à sévère, touchant souvent les garçons plus gravement.
8. Quels sont les risques associés à l’obésité dans le syndrome d’Alström ?
L’obésité peut entraîner des problèmes métaboliques tels que le diabète de type 2 et augmenter le risque de cardiomyopathie dilatée.
9. Y a-t-il des traitements spécifiques pour la cardiomyopathie dilatée ?
Oui, les patients peuvent nécessiter un traitement avec des inhibiteurs de l’enzyme de conversion pour gérer leur condition cardiaque.
10. Quelle est l’importance du suivi médical ?
Un suivi régulier est crucial pour surveiller l’évolution des symptômes et adapter le traitement en fonction des besoins du patient.
🌐 Sources
- has-sante.fr – Syndrome d’Alström – PNDS 2019
- sante-sur-le-net.com – Syndrome d’Alström : définition, symptômes et traitements
- orpha.net – Syndrome d’Alström
- researchgate.net – Le syndrome d’Alström : une cause exceptionnelle d’hypogonadisme primitif
- thpanorama.com – Síndrome de Alström: Síntomas, Causas, Tratamento
- caducee.net – La cause génétique du syndrome d’Alström a été identifiée